
Accelerating the Bioactivity Frontier with AI Prediction
From possibilities to breakthrough — faster than ever before.
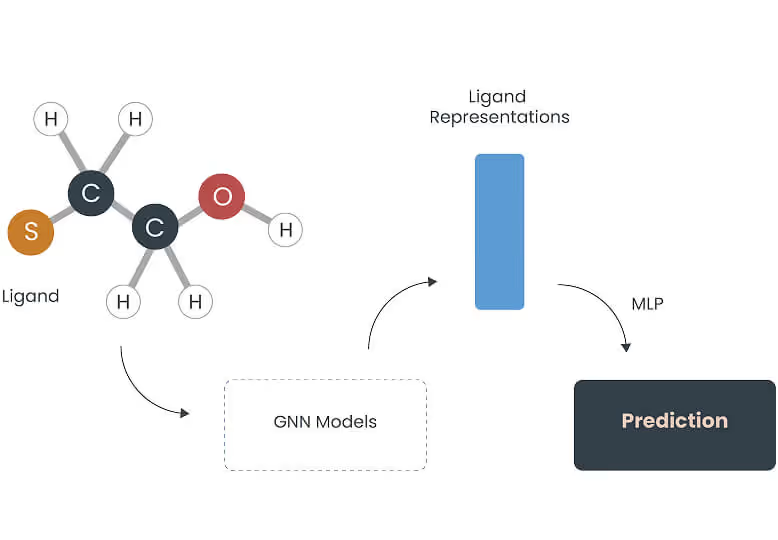
At NYB, we believe the next generation of therapies is hidden within nature’s vast, unexplored chemical spaces. To speed up the discovery process, we have developed successive proprietary AI models predicting bioactivity with an unwavering focus.
At the same time, we eliminate disjointed research silos by integrating high-performance tools into a single, streamlined workflow.
A Different Approach
Vecura, our integrated compound discovery platform, deploy a continuum of over 200 AI models, including our proprietary models to predict bioactivity levels: Drug-Target Interaction Graph Network (DTIGN), Ligand-Protein Space Mapping (LigoSPACE), and Binding Affinity and Interaction Network Design (BIND).
Leveraging these models and a unified workflow, Vecura reduces the time required to identify hit compounds by up to 68% for a client relative to traditional approaches:
- We map how molecules interact, not just how they look
- We identify where binding actually occurs, not just where it might
- We simulate real biological environments, not simplified models
Proprietary Models, Measurable Impact
This approach is grounded in measurable performance. In a paper published in the IEEE Journal of Biomedical and Health Informatics, our Drug-Target Interaction Graph Network (DTIGN) model has demonstrated:
Compound Discovery Innovation
NYB continues advancing our AI model capabilities. Evolving from the Drug-Target Interaction Graph Neural Network (DTIGN), it progressed to LigoSPACE (DTIGN 2.0) for multi-site protein mapping. The latest Bioactivity-driven Interaction Discovery or BIND (LigoSPACE 2.0) builds upon iteration now integrates contrastive learning and uncertainty-weighted consistency.
2024
DTIGN incorporating graph neural network etc.
Dec 2025
LigoSPACE (DTIGN 2.0) mapping multiple pockets across the entire protein.
Apr 2026
BIND (LigoSPACE 2.0) incorporating contrastive learning etc.
Introducing DTIGN
We move beyond approximation—toward predictive clarity grounded in real biological interactions.
Our DTIGN framework has demonstrated superior performance across bioactivity prediction benchmarks, outperforming nine leading non-interaction-based methods by an average improvement of 27.03%.

LigoSPACE uses three innovative approaches
LigoSPACE precisely measures the "unfilled pockets" within protein binding sites at the atomic level. This reveals whether a drug molecule truly fills the space it needs to, or if there are gaps that could let interfering molecules slip through.
Why it matters: It's the difference between a drug that looks like it fits and a drug that actually fits perfectly.
Instead of analyzing each binding site in isolation, LigoSPACE maps multiple pockets across the entire protein. This gives a complete view of where a drug will interact, not just a narrow snapshot.
Why it matters: Drugs don't work in isolation—they interact with multiple sites. Seeing the full landscape predicts real-world outcomes better.
Most AI systems use a basic mathematical approach to compare candidates. LigoSPACE uses a more sophisticated method that captures the relationships between different drug candidates, which is what actually matters in drug discovery.
Why it matters: It mimics how scientists actually think about drugs: "Is this candidate better than that one?"—not just absolute numbers.

BIND builds upon LigoSPACE research
The BIND AI model learns to distinguish between:
• Meaningful interactions — The aspects of a drug molecule that truly determine whether it works
• Noise or coincidence — Random patterns that happened to appear in the training data but don't actually matter
Why it matters: It's the difference between a drug that looks like it fits and a drug that actually fits perfectly.
The BIND AI model learns by comparing:
• Similar drug molecules — Pulling them together in its reasoning to understand what they have in common
• Different drug molecules — Pushing them apart to understand what makes them fundamentally differentThis reinforces learning about the deeper structural relationships that actually matter, not just surface-level patterns.
Why it matters: It prevents the AI from memorizing superficial patterns and forces it to understand deeper principles. These deeper principles remain valid even when conditions change.
Here's the innovation: The BIND AI model adapts to new conditions using only new data, without needing to access or share the original training data.
This is like teaching someone to fish using only observations of the current location, without revealing your private fishing spot or how you originally learned to fish.
Why it matters: Companies can improve AI predictions for their specific research without revealing proprietary information. Competitive advantage is protected while predictions improve.

Expanding the Boundaries of Possibility
Advancing Therapeutics
- Oncology
- Metabolic diseases
- Cardiovascular conditions

Translating Discovery into Preventive Health
- Identification of high-potential natural compounds
- Scientific validation of their biological activity
- Development of evidence-based nutraceutical solutions
